Genome Assembly
Cole Lyman
19 October 2016
What is Genome Assembly?
Genome assembly is often compared to putting a puzzle together. In this analogy, the pieces of the puzzle are individual reads that we get from a DNA sequencer. The ultimate goal of putting a puzzle together is to find out where each and every piece fit exactly, this way you can see the completed picture. While this is also true of genome assembly, we need to be realistic. The first step in genome assembly is to generate contigs, or in the words of our puzzle analogy, to combine pieces that we know go together.
What is a Contig?
A contig is short for contiguous sequence. It is a sequence that is longer than the reads, and shorter than the genome (technically the whole assembled genome could be considered a contig in an organism with a single chromosome, but for our purposes it is considered to be shorter than the entire genome). If you have a puzzle that is a picture of a beach and a ocean, you would combine all of the tan colored pieces and then all of the blue colored pieces. You may even combine the pieces that make up the colorful beach umbrellas littering the beach. When you do this you are generating contigs! You are making small chunks of the whole because they are easy to identify, and it is the same with genome assembly.
How Do We Discover Contigs?
Well, we know that the pieces of our puzzle are reads. The regular puzzle piece has four sides with different shapes that define exactly where it should go. Our reads also have “sides” that tell us the proper place it should be in the contig. These “sides” are revealed when we break the read up into kmers and inserting them into a de Bruijn graph. We then can construct the contigs by continuing the non-branching nodes (a node that has exactly one incoming edge and one outgoing edge).
Imagine that we are assembling an incredibly small genome, with way too few reads, which are the following:
ACTGT
TCTGT
CTGTT
CTGTA
GCATA
TGTTA
GTTAC
The reads are of length 5. The de Bruijn graph using the kmer length of 5 (the entire read) for this set of reads would look like this (the non-branching node is in red):
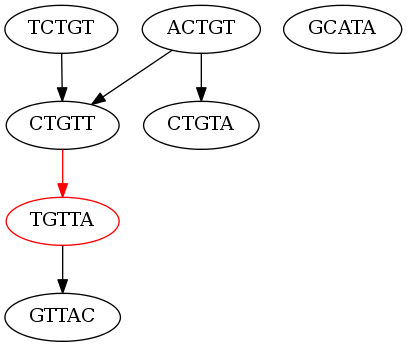
As you can see there is only one non-branching node in the graph. We can only generate the following contigs:
ACTGT
TCTGT
CTGTTA
CTGTA
GCATA
TGTTA
GTTAC
I hope it is clear that this is not a very good assembly. Why isn’t this assembly good? Well for the most part all of our contigs are the same length of our reads, except for one, which is only one more base pair than the read length. This assembly is equivalent to only connecting one puzzle piece. Let’s see how we can improve this assembly.
Kmer Length
The length of kmer that you use to construct your de Bruijn graph will greatly influence your assembly, for better or for worse. What will happen if we decrease the kmer length from 5 to 3?
Here is the de Bruijn graph of kmer length 3:
Here is the de Bruijn graph of kmer size 3:
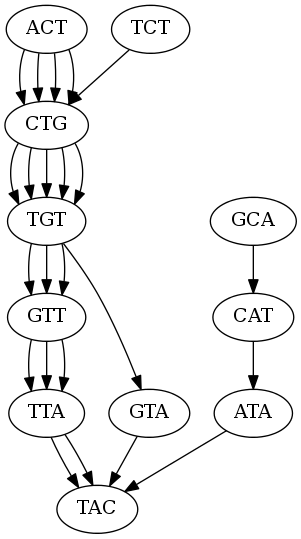
What is up with all of the edges?
Each edge represents an occurance of that kmer, for example the kmer ACT has 4 edges to CTG because there are 4 occurances of CTG.
Let’s clean this graph up by giving weights to the edges.
The number on each edge represents how many times that edge is repeated.
Here is the de Bruijn graph of kmer size 3 with weighted edges (the non-branching nodes are in red):
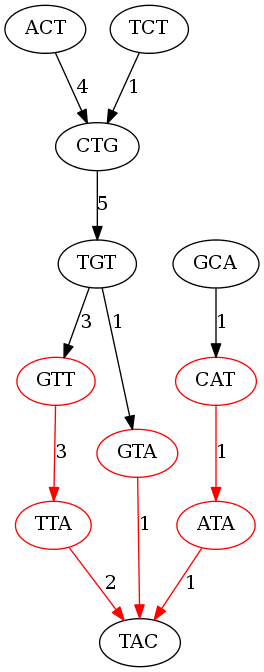
Let’s see if a kmer length of 3 is any better; we definitely have more branhcing nodes, but will that lead to longer contigs? Essentially we have made our puzzle pieces smaller, so just because we can put together more puzzle pieces doesn’t mean that we are building more of the puzzle. Here are the contigs generated by the graph of kmer length 3:
ACT
TCT
CTG
TGTTA
TGTA
GCATA
TAC
Well, compared to the assembly using kmers of length 5 we have the same number of contigs, with a shorter average, and the longest contig is only the length of the reads. Is this assembly better or worse than the last one? Short answer, yes. You have to define “better” when classifying assemblies. You may want to have the longest average contig length, or simply the longest contig. You may have some other metric like N50 in which you determine which assembly is “better.”
Filtering Errors
One way of accounting for errors in your algorithm is to remove edges below a certain weight. When an edge has a higher value, it occurs more often. If an edge occurs more often, then it is more likely to be a valid contig.
What is Next?
You may be asking yourself, “I can see how we generate contigs, and how that can be useful, but we still don’t have one sequence that represents the genome. We haven’t fully completed the puzzle. How do we do that?” If you would like to fully complete the puzzle, to put all of these contigs together, you would have to construct scaffolds. A scaffold is a representation of how contigs relate to each other as well as accounting for gaps in the sequence. Depending on how much data you have, you may not be able to create one continuous sequence.
NOTE: Scaffolding is not required for the CS 418/BIO 365 Genome Assembler project. Generating contigs is good enough!